恶性肿瘤是常见的严重威胁人类生命和影响人类生活质量的重大疾病之一。探索肿瘤细胞中普遍存在但又异于正常细胞的生物学特性,并针对该特性进行特异性干预,是提高肿瘤治疗疗效的关键。肿瘤细胞代谢的改变是肿瘤的重要特征之一,其与肿瘤的发生发展互为因果。随着对肿瘤代谢分子生物学基础研究的深入,越来越多的证据表明,代谢过程的改变包括但不局限于代谢酶自身的突变或代谢调控蛋白的活性变化,均可导致肿瘤细胞的代谢重编程,使肿瘤细胞具有特征性的代谢模式。翻译后修饰是细胞内的特定化学基团从一个蛋白传递到另一个蛋白的反应,是细胞信号转导的重要环节。肿瘤细胞通过翻译后修饰介导原癌基因的激活和/或抑癌基因的抑制,弱化细胞周期调节和增强增殖生长信号,促使肿瘤发生和快速发展。
肿瘤细胞如何通过翻译后修饰介导代谢重编程,为肿瘤快速增殖提供代谢优势,翻译后修饰所调控的代谢特征是否能够成为预测肿瘤发生发展的生物标志物并成为靶向治疗肿瘤的新靶点,正成为近年来的研究热点领域。为了进一步了解这一领域的研究进展,本文集中介绍了近年来不同类型的翻译后修饰参与肿瘤代谢重要途径重编程的主要研究成果。
万世万物,此消彼长,新陈更迭,皆可以被认为是宏观意义上的代谢。从微观的角度出发,代谢是指生物体内为维持生命而发生的所有生物化学反应的总和。细胞代谢涵盖了细胞内发生的所有化学过程,以维持体内稳态并总体上确定其能量状态,包括合成代谢和分解代谢。合成代谢是指由较简单的成分合成复杂分子并储存能量,而分解代谢是把复杂分子分解成更简单的成分而释放能量。细胞代谢对于生命有机体的生长、繁殖、结构维持,应激等分子生理机制至关重要,因此,细胞代谢整个过程也受到严密精准的调控。
翻译后修饰在细胞代谢过程的精密调控系统中常常作为“分子开关”,起着“四两拨千斤”的作用。翻译后修饰是指蛋白质在翻译后经历的一个共价加工过程,即通过1个或几个氨基酸残基加上修饰基团或通过蛋白质水解剪去基团而改变蛋白质的性质。蛋白翻译后修饰能够在生理及病理条件下,通过影响蛋白的活性、稳定性、定位以及信号转导等,快速调节功能蛋白的活化、转位、组装等过程,扩大蛋白功能的多样性。翻译后修饰类型包括经典的磷酸化、泛素化、甲基化修饰,以及新近发现的糖基化、乙酰化、丙二酰化、琥珀酰化修饰,以及乳酸酰化等。代谢重编程(metabolic reprogramming)是肿瘤发生发展的十大特征之一,也是近年再次受到关注的热点研究方向。
肿瘤细胞通过代谢重编程提供生长、增殖、免疫逃逸和转移所需要的充足能量、必需大分子原料、适宜的氧化还原平衡环境。在此过程中,肿瘤细胞也表现出高度的代谢可塑性,而这种可塑性很大程度上是通过翻译后修饰来得以实现。
1 翻译后修饰与肿瘤细胞葡萄糖代谢通路重编程
1.1 翻译后修饰与肿瘤细胞糖酵解通路
代谢改变在肿瘤发生发展中的作用并非是一个崭新的发现,早期的肿瘤生物学研究就已经意识到葡萄糖代谢模式的改变是肿瘤组织区别于正常组织的一个重要特征。葡萄糖经细胞表面葡萄糖转运蛋白摄取进入细胞胞浆后,再经糖酵解生成丙酮酸,当氧供应充足时,丙酮酸进入线粒体并发生有氧氧化,最终转化为CO2和H2O,并产生大量三磷酸腺苷(adenosine triphosphate, ATP);在氧供应不足或不能被充分利用时,丙酮酸不进入线粒体,而是被细胞质中乳酸脱氢酶A(lactic dehydrogenase A, LDHA)还原成乳酸,同时产生少量的ATP。早在近100年前,德国科学家Otto Warburg发现肿瘤细胞即使在有氧情况下,也主要通过糖酵解模式进行糖代谢以提供能量,这种有氧糖酵解的代谢方式随后被命名为瓦伯格效应(Warburg effect),呈现出高葡萄糖消耗、低ATP合成和高乳酸生成的特点。肿瘤细胞实现代谢模式从氧化磷酸化到糖酵解通路的转变也是肿瘤发生发展的关键因素。肿瘤细胞一方面在转录水平上调控葡萄糖代谢通路中关键代谢酶的表达水平,同时通过多种翻译后修饰调节关键代谢酶的活性、组成、亚细胞定位等生物学特性来实现代谢方式重编程。
1.1.1 葡萄糖转运蛋白
葡萄糖转运蛋白(glucose transporters, GLUT)是一类负责机体葡萄糖进出入组织器官的关键门控蛋白,专职负责组织器官的能量供给和机体葡萄糖水平稳态调节功能。葡萄糖转运蛋白是细胞利用葡萄糖的重要环节,是调节细胞葡萄糖使用的第一步。GLUT1是一类广泛分布于众多组织器官的葡萄糖转运蛋白,对于维持人的正常生理功能极为重要,其表达和功能异常与肿瘤细胞的发生发展具有重要的意义[2]。大量研究表明,GLUT1蛋白在多种肿瘤类型细胞中高度表达,提示与肿瘤细胞快速增长需要大量葡萄糖密切相关。尽管目前还没有数据说明翻译后修饰直接作用于GLUT1蛋白本身,影响其转运葡萄糖的功能与效率,但有研究表明持续激活形式的AKT能够促进FL5.12细胞的GLUT1转移到细胞膜上,提高葡萄糖的摄取和细胞内ATP水平以及糖酵解速率,并与该细胞的肿瘤发生密切相关。在多种类型肿瘤细胞中高表达的AKT 作为丝氨酸/苏氨酸激酶,是生长因子介导的细胞存活机制中的重要效应分子并在肿瘤的发生发展中发挥着极其重要的作用。这些研究显示AKT信号通路与GLUT1蛋白的细胞膜转位、葡萄糖摄取增加和肿瘤代谢模式的转变有着潜在的联系。但是, AKT是直接作用于GLUT1 本身还是通过其他蛋白来介导GLUT1蛋白的细胞膜转位还期待进一步研究。
1.1.2 己糖激酶
己糖激酶(hexokinase,HK)作为糖酵解通路中第一个代谢酶,负责把葡萄糖转化成为六磷酸葡萄糖(G-6-P)。人己糖激酶有4种同源异构体,且组织丰度、与底物结合力各有不同。HK1 主要表达在脑组织中,HK2表达于心肌、脂肪等组织中,HK3 广泛表达于各种组织,而HK4 的表达局限于肝和胰腺。 HK1、HK2 以及 HK3与葡萄糖的结合能力约为HK4 的250倍[3]。研究表明c-Src在Y732位点上磷酸化HK1,促进肿瘤细胞的糖酵解速率以及其增殖、侵袭和转移能力[4]。在肿瘤发生发展过程中,原本表达的低活性的HK4被高活性的HK2 取而代之,从而适应肿瘤细胞快速消耗葡萄糖的需要。事实上,肝细胞癌中HK2的确高表达,并且 HK2 的表达水平与肿瘤恶性程度和致死率呈正相关。虽然在正常脑组织中HK1优势表达,但在乳腺癌脑转移的组织中以及胶质瘤中,HK2高表达并提示预后不良。因此,HK2在肿瘤中的高表达被认为是肿瘤进行代谢重编程而适应肿瘤快速生长需要的结果。此外,早先在心肌细胞和成纤维细胞中的研究显示AKT直接在Thr473位点上磷酸化HK2,磷酸化修饰的HK2转位到线粒体中,与线粒体外膜蛋白VDAC结合,阻止细胞色素C的释放和细胞凋亡的发生。肿瘤细胞HK2的翻译后修饰对其功能影响和在肿瘤发生发展中的作用也受到广泛关注。在乳腺癌细胞中,HK2能够被激酶PIM2在Thr473位点磷酸化,该位点的磷酸化能增强HK2蛋白的稳定性和酶活性,促进乳腺癌细胞糖酵解和肿瘤生长,并促进其对紫杉醇的耐药[5]。在肠癌细胞中,AKT2可能是导致HK2 Thr473磷酸化的上游激酶,并促进HK2的酶活性和肠癌细胞的侵袭和转移能力[6]。在前列腺癌细胞中,HK2可以在K315和K492上被SUMO化。SUMO特异蛋白酶SENP1介导HK2的去SUMO化。SUMO缺陷后,HK2优先与线粒体结合,并同时增加葡萄糖消耗和乳酸生成并降低线粒体呼吸。这种代谢重编程支持前列腺癌细胞的增殖并保护细胞免受化学疗法诱导的细胞凋亡的影响。以上研究提示肿瘤细胞中HK2的表达、功能、亚细胞定位可以被多种翻译后修饰所调节,所引发的代谢重编程在肿瘤发生发展、侵袭浸润、耐药性产生等肿瘤分子病理过程中发挥着重要作用[7]。
1.1.3 磷酸果糖激酶
磷酸果糖激酶1(phosphofructokinase-1, PFK1)是糖酵解过程的主要限速酶,在ATP与Mg2+参与下,作用于6-磷酸果糖并生成1,6-二磷酸果糖,这是糖酵解途径的第二次磷酸化反应。在低氧环境下,肿瘤细胞PFK1在Ser529位点发生糖基化,抑制PFK1活性,降低了糖酵解的速率和乳酸的生成,将糖代谢转向磷酸戊糖途径,产生更多的还原物质NADPH。看似与肿瘤细胞糖酵解普遍增强观念相矛盾的结果,提示肿瘤细胞不仅利用糖酵解来生成大量的ATP为其快速增殖提供能量,也提供大量的代谢中间产物为生物合成途径如磷酸戊糖途径提供原材料,来满足肿瘤细胞快速增殖的需要[8]。此外,肿瘤细胞还利用磷酸戊糖途径所产生的还原物质NADPH以中和快速增殖过程中所产生氧自由基来抵抗自由基所引发的肿瘤细胞损伤和凋亡[8]。PFK1的同源异构体 PFKP 可以被AKT在S386 上磷酸化,该磷酸化修饰抑制了PFKL的降解,从而导致了PFKL蛋白水平的积聚,促进了胶质瘤细胞有氧糖酵解和肿瘤的生长[9]。磷酸果糖激酶2是一个有趣的双功能蛋白,在非磷酸化状态下具有激酶的功能,磷酸化6-磷酸果糖,生成2-6双磷酸果糖。而在磷酸化的状态下,其转变为磷酸酶,催化2-6双磷酸果糖转化为6-磷酸果糖。2-6双磷酸果糖是磷酸果糖激酶1的强力激动剂。因此,磷酸果糖激酶-2/2-6二磷酸果糖磷酸酶-2,(PFK-2/FBPase-2)是其更为完整准确的名称[10]。PFK-2/FBPase-2有4种同工酶,PFKFB1-4,在不同肿瘤的不同阶段发挥着不同的作用。在BRAF突变的黑色素瘤中,RSK直接磷酸化PFKFB2,促进 PFKFB2的活性,提高糖酵解通路水平,加速了BRAF突变的黑色素瘤的生长[11]。PFKFB3在K472位点能够被乙酰化,该修饰削弱了PFKFB3的核定位信号,并使其滞留在细胞质当中。而细胞质中的PFKFB3能够被AMPK进一步磷酸化,从而导致PFKFB3的激活,并进一步激活糖酵解通路, 揭示了代谢酶PFKFB3的乙酰化修饰对糖酵解和肿瘤化疗敏感性的重要调控作用,提示通过靶向抑制PFKFB3而提高顺铂等化疗药物的敏感性有可能成为临床上一个新的治疗策略[12]。
1.1.4 磷酸甘油酸变位酶
磷酸甘油酸变位酶(phosphoglycerate mutase, PGAM)在糖酵解通路中负责将3-磷酸甘油(3-PG)转化为2-磷酸甘油(2-PG),是协调糖酵解,磷酸戊糖途径和色氨酸合成途径的关键酶,在肿瘤细胞的代谢重编程中也扮演着重要的角色。有研究发现在肿瘤细胞中PGAM1的Y26位点普遍被多种酪氨酸激酶磷酸化,该位点的磷酸化通过增强活性形式的PGAM1的稳定性提高其活性,调节其底物3-PG和产物2-PG的比例,协调糖酵解通路与合成代谢通路,为肿瘤快速增长提供优势。PGAM1的K251/253/254位点的乙酰化能够增强乳酸生成,SIRT1是以上位点的去乙酰化酶,从而调控有氧糖酵解速率。但是乙酰化PGAM1是否推动肿瘤代谢模式的转变重要因素,以及乙酰化PGAM1在肿瘤发生发展过程中的作用目前尚不完全清楚。
1.1.5 丙酮酸激酶M2 型
丙酮酸激酶M2 (pyruvate kinase M2, PKM2)型是广受关注的代谢酶之一,它是糖酵解通路中最后一个限速酶,负责将磷酸烯醇丙酮酸转化为丙酮酸。根据丙酮酸激酶在不同组织中的分布,丙酮酸在哺乳细胞内有4种类型,分别是PKL、PKR、PKM1和PKM2。PKL主要分布在肝脏、肠、胰腺以及肾脏,PKR主要分布在红细胞中。PKM1和PKM2是同一基因不同的剪切体,PKM1在需要大量能源供应的组织和器官中表达普遍上调,如心脏、脑组织、肌肉,而PKM2则在所有增殖细胞中特别是在肿瘤和胚胎组织中表达。PKM1与PKM2的酶活性质和调节方式不同。PKM1是持续激活形式,与底物磷酸烯醇丙酮酸有着很强的结合力;而PKM2则受到复杂的别构调控,对肿瘤的生长和进展发挥着至关重要的作用。在正常组织中,PKM2蛋白在高活性的四聚体形式和低活性的二聚体形式转换。在肿瘤组织中,PKM2蛋白倾向于以低活性的二聚体形式,从而积聚PKM2上游的代谢中间产物作为前体物质通过磷酸戊糖途径为合成肿瘤快速生长和增殖所必需的生物大分子提供原材料。众多形式的翻译后修饰包括磷酸化、乙酰化、琥珀酰化等参与调节PKM2活性和功能,通过促进细胞代谢重编程、血管生成、肿瘤转移和基因组不稳定性推动肿瘤的发生和进展。首先,多种酪氨酸激酶包括成纤维细胞生长因子(FGFR1)在Y105位点直接磷酸化PKM2, 阻碍辅助因子1,6-二磷酸果糖与PKM2的结合,并抑制高酶活性的四聚体形成。 PKM2 Y105位点磷酸化在一系列肿瘤细胞中显著升高,对肿瘤细胞代谢模式向有氧糖酵解转变和快速增殖发挥着重要作用。在高糖环境下,p300/CBP相关因子(PCAF)在K305位点乙酰化PKM2,降低其与底物PEP的结合,抑制其酶活性并促进其通过分子伴侣介导的自噬和溶酶体依赖的降解。外源性表达模拟乙酰化的PKM2 K305Q可以导致糖酵解中间产物的聚集,促进肿瘤的增殖和生长。在低氧或氧自由基存在的环境下,PKM2 C358位点发生氧化并导致酶活性降低,将代谢产物G-6-P转向磷酸戊糖通路,提高重要还原物质 NADPH的合成,使肿瘤细胞避免氧化应激带来的损伤。
以上研究显示PKM2被修饰后导致酶活性降低,聚集代谢中间产物以满足肿瘤细胞对生物合成原料和抵抗氧化应激损伤的需求。另有研究表明JNK-1磷酸化PKM2 Thr365位点,提高PKM2酶活性,减少还原当量GSH水平,促进肝癌细胞系凋亡,但其中的具体分子机制和临床意义,仍需进一步研究。肝癌细胞系中存在大量PARP14,抑制JNK-1对PKM2 Thr365的磷酸化和激活,促进肝癌细胞瓦博格效应的产生[18]。PKM2的翻译后修饰不仅可以调节其酶的活性,也可以调节其在细胞内的定位。PKM2在S37的位点上被ERK2磷酸化,该修饰促进PKM2转入进细胞核,行使非代谢酶功能,作为蛋白激酶在T11位点磷酸化组蛋白H3,上调细胞周期蛋白和原癌基因的表达,从表观调控水平上促进肿瘤细胞的增殖[19-20]。PKM2 S37磷酸化水平还受磷酸酶cdc25A 去磷酸化调节参与调控PKM2蛋白激酶功能和肿瘤细胞瓦博格效应的形成[21]。在营养压力如低糖状态下,PKM2 K433位点的琥珀酰化介导PKM2进入线粒体,抑制线粒体VDAC3的降解,提高线粒体膜的通透性,产生更多的ATP,协助细胞在营养缺失下生存[22]。PKM2在Arg445/447/455位点上被CARM1(PRMT4)甲基化修饰,定位于线粒体相关ER膜上,控制钙离子的转运和氧化磷酸化水平,促使肿瘤细胞更加依赖于有氧糖酵解通路,导致瓦博格效应的产生[23]。HAUSP (herpesvirus-associated ubiquitin-specific protease) 可以与PKM2结合,并可能是其潜在的去泛素化酶,但HAUSP 对PKM2的去泛素化作用在肿瘤发生发展和在肿瘤代谢重编程中的意义有待于进一步探索。
1.1.6 乳酸脱氢酶
乳酸脱氢酶(lactate dehydrogenase, LDH)大多数是由LDHA,LDHB这两种亚基通过不同组合方式组成的5种同/异四聚体(A4、A3B1、A2B2、A1B3、B4)分别称之为LDH 1~5。乳酸脱氢酶在糖酵解通路的终端,以NADH为辅助因子,负责催化丙酮酸还原生成乳酸,并且氧化NADH为NAD+。在正常增殖细胞中,绝大部分丙酮酸会进入线粒体,通过丙酮酸复合体转化为乙酰辅酶A,参与三羧酸循环和氧化磷酸化。而在肿瘤细胞和快速增殖的细胞中,葡萄糖摄取以及相对应的丙酮酸水平增加,进入线粒体的丙酮酸并没有相应增多,乳酸脱氢酶在转录因子HIF和Myc调控下表达增加,加速了丙酮酸向乳酸的转化。肿瘤细胞中不同类型的翻译后修饰也参与调控LDHA的蛋白水平和活性,促进乳酸的产生。有研究显示在肿瘤细胞中普遍高表达的酪氨酸激酶受体包括FGFR1、FLT3-ITD、BCR-ABL、JAK2能够在Y10位点直接磷酸化LDHA,协助形成具有高活性四聚体、激发酶的活性、促进丙酮酸向乳酸的转化、并调节NADH/NAD+的平衡,促进肿瘤细胞的增殖和生长。乳酸脱氢酶Y10 磷酸化水平在多种肿瘤细胞系,包括肺癌、白血病、头颈部肿瘤、乳腺癌和前列腺癌等癌细胞均明显升高,也与促进乳腺癌侵袭与转移密切相关,提示其可能成为肿瘤发生发展以及转移的生物标志物[24-25]。在胰腺癌中,LDHA在K5位点发生乙酰化,抑制其酶的活性,同时也被热休克蛋白HSC70所识别和介导降解,下调LDHA蛋白水平。早期胰腺癌组织LDHA K5乙酰化水平与癌旁组织相比明显降低,提示LDHA K5乙酰化可能与胰腺癌的发生有关[26]。另外,LDHA K222位点的棕榈酰化提示胃癌预后不良,可能是由于棕榈酰化的LDHA溶酶体降解通路被削弱,从而导致LDHA蛋白水平升高,并与胃癌的侵袭和转移也密切相关[27]。
1.2 翻译后修饰与肿瘤细胞三羧酸循环
1.2.1 丙酮酸脱氢酶复合体
肿瘤细胞以及快速增殖的细胞中,葡萄糖摄取增加,所生成的丙酮酸也相应增多,丙酮酸位于糖酵解和TAC循环的交叉点,其代谢命运主要受乳酸脱氢酶和丙酮酸脱氢酶复合体(pyruvate dehydrogenase complex, PDC)调控。前者通过将丙酮酸转化为乳酸来促进糖酵解,而后者通过将丙酮酸催化为乙酰辅酶A来参与和维持TAC循环。肿瘤细胞中进入线粒体参与三羧酸循环的丙酮酸与糖摄取的增加不成比例,这提示肿瘤细胞线粒体功能部分受损可能是导致瓦博格效应的因素之一。丙酮酸脱氢酶复合体是位于线粒体内负责将丙酮酸不可逆地转变为乙酰辅酶A,进入三羧酸循环的多酶复合物。丙酮酸复合体首要成分是丙酮酸脱氢酶(PDHA 或PDH E1),其活性受磷酸化和去磷酸化来实现的。丙酮酸脱氢酶激酶(PDK或PDHK)在S293等位点上磷酸化PDHA,并抑制其活性,而磷酸酶PDP1则去除这些位点上的磷酸化,激活PDHA活性。研究发现PDHK1的Y136、Y243、Y244位点被FGFR1等酪氨酸激酶磷酸化,Y136的磷酸化促进PDHK与丙酮酸复合体E2和底物PDHA的结合,Y243/Y244磷酸化增强了PDHK1与ATP的结合,提高其激酶活性,磷酸化PDHA1,从而进一步抑制PDHA的活性,调节丙酮酸的转化效率。除了在Ser293位点上受PDHK磷酸化调控外,PDHA也受酪氨酸激酶磷酸化修饰和直接调控,PDHA Y301磷酸化可以降低其与底物丙酮酸的结合,减少丙酮酸的转化。这些研究展示出,同一个蛋白通过不同位点的磷酸化修饰,以不同的分子机制协同调控PDC活性,促成瓦博格效应的形成。PDP1 Y94 被FGFR1、JAK2、BCR-ABL等酪氨酸激酶磷酸化抑制其与PDC E2的结合,从而进一步削弱对PDHA磷酸酶活性。PDP1与PDHA一样,也存在多位点磷酸化。PDP1 Y381 磷酸化引发去乙酰化酶SIRT3脱离PDC中心,招募乙酰转移酶ACAT1至PDC中心并乙酰化PDP1 K202位点以及PDHA K321 位点,重构PDC中心蛋白组成架构,抑制PDC活性,促进肿瘤细胞增殖和生长[30]。以上研究展示了肿瘤细胞利用多种翻译后修饰形式在不同层面和不同位点协同调控PDC的中心成分和功能,对细胞代谢进行重编程,减少丙酮酸在线粒体的利用,增加其进一步转化为乳酸,以满足肿瘤细胞快速增殖的需求。与原发性肿瘤相比,转移性肿瘤中AMPK活性富集, 并直接触发PDHA在Ser295和Ser314上的磷酸化,PDHA Ser314磷酸化消除了PDHA和PDHK之间的相互作用,解除了PDHK对PDHA的负性调控,PDHA的Ser295和Ser314过度磷酸化通过提高PDH的活性,将肿瘤新陈代谢重新导向TCA周期,从而保护已扩散的癌细胞免受代谢和氧化应激诱导的细胞死亡,并促进癌症转移[31]。另有研究显示,PDC的主要蛋白成分,包括PDHA、 PDP1,其在EGF或血清处理后可以进入细胞核内,并以核内的丙酮酸为原料合成乙酰辅酶A,作为组蛋白乙酰化的重要供体,促进肿瘤细胞组蛋白乙酰化和周期调控,展现了线粒体蛋白在线粒体-细胞核不同细胞器之间穿梭发挥相关功能,“跨界”调控表观遗传,促进肿瘤细胞增殖与分化的模型。但是,线粒体蛋白如何进入细胞核内,是否存在其他翻译后修饰调控从线粒体转位到细胞核这一过程,还有待于进一步研究[32]。
1.2.2 异柠檬酸脱氢酶
异柠檬酸脱氢酶(isocitrate dehydrogenase, IDH)是三羧酸循环中重要的酶,其主要功能是将异柠檬酸转化成草酰琥珀酸再进一步氧化成 α-酮戊二酸(α-KG)。此外,IDH 提供的还原型烟酰胺腺嘌呤二核苷酸磷酸(NADPH)能够用于细胞中脂类物质的合成参与不饱和脂肪酸的降解过程,用于体内抗氧化作用,保护机体免受氧化损伤,延缓衰老过程。IDH 家族主要包括3 种酶,分别为IDH1、IDH2和IDH3。IDH1蛋白主要存在于过氧化氢酶体及细胞质中,而IDH2 蛋白主要存在于线粒体中,但IDH3 基因的位置尚未确定。大多数IDH 为同源二聚体,每个亚基均有一个活性中心。IDH 突变和肿瘤的发生有着紧密关联,在AML、胶质瘤和软骨肉瘤等恶性肿瘤中存在30%~80%的突变率,而普遍的突变为 IDH1 基因R132C位点突变,其次是IDH1基因R132H位点突变等,突变位点均位于IDH酶的活性位点上,因而突变对酶的活性有直接影响。此外,异柠檬酸脱氢酶 (IDH) 活性位点突变导致新形态酶活性,导致形成的D-2-羟基戊二酸(D-2HG),通过抑制参与组蛋白和DNA去甲基化的酶来改变细胞的表观遗传状态,从而推动多种组织类型癌症的形成。有研究显示两组不同的酪氨酸激酶之间存在相互作用,分别在Y381 和Y42位点磷酸化野生型和突变型的IDH1,通过促进辅助因子或底物结合到IDH1,或促进高活性二聚体形成等机制激活野生型和突变型IDH1,保障肿瘤细胞的快速生长和增殖。此外,线粒体定位的IDH2 R140Q 突变体通常伴有K413的乙酰化,通过抑制高活性IDH2的二聚化和辅助因子NADPH的结合,负性精细调控IDH2活性,将内在过高的2-HG水平调控在优化区间内,一方面促使AML细胞的转化,另一方面也避免过高2-HG水平对肿瘤细胞的毒性作用[34-35]。
1.3 翻译后修饰与肿瘤磷酸戊糖途径
磷酸戊糖途径(pentose phosphate pathway)也称为单磷酸己糖支路(hexose monophosphate shunt),是葡萄糖-6-磷酸经代谢产生NADPH和核糖-5-磷酸的途径,主要在细胞质中进行,为合成代谢提供多种原料,如为脂肪酸、胆固醇的生物合成提供NADPH;为核苷酸辅酶、核苷酸的合成提供5-磷酸核糖。葡萄糖6磷酸脱氢酶,(glucose-6-phosphate dehydrogenase, G6PD) 是磷酸戊糖途径第一个限速酶,研究发现在不同器官来源的肿瘤细胞中G6PD发生多种类型的翻译后修饰调控G6PD功能。G6PD上的S 84位点上的糖基化,SIRT2介导的K403位点去乙酰化,PLK1介导的G6PD T406/T466磷酸化,不同程度地激活G6PD,促进葡萄糖进入磷酸戊糖途径,增加Ru-5-p, NADPH的产出,促进肿瘤细胞的生长和增殖。6-磷酸葡萄糖酸脱氢酶(6-phosphogluconate dehydrogenase 6PGD) 是PPP通路上的第三个重要酶,研究发现6PGD分别在K76 和K294位点在乙酰转移酶DLAT和ACAT2作用下发生乙酰化,在Y481位点被SRC家族激酶FYN磷酸化,K76位点的乙酰化和Y481的酪氨酸磷酸化促进辅助因子NADP+与6PGD结合,K294位点的乙酰化促进高活性6PGD二聚体的形成,从而进一步激活6PGD和PPP通路,产生更多的核糖-5-磷酸和NADPH,用于肿瘤细胞原料核酸合成和抵抗氧化自由基损伤。此外6PGD的产物5-磷酸核糖(Ru-5-P)通过破坏活性LKB1复合物抑制AMPK活化,从而活化乙酰辅酶A羧化酶1和脂肪形成,促进细胞增殖和肿瘤的生长。以上研究展示了磷酸戊糖途径中不同类型的翻译后修饰调节代谢酶的活性,为肿瘤细胞的生长增殖提供大分子原料,或/和通过该通路中的代谢产物作为信号分子直接参与影响肿瘤细胞增殖的信号转导通路。
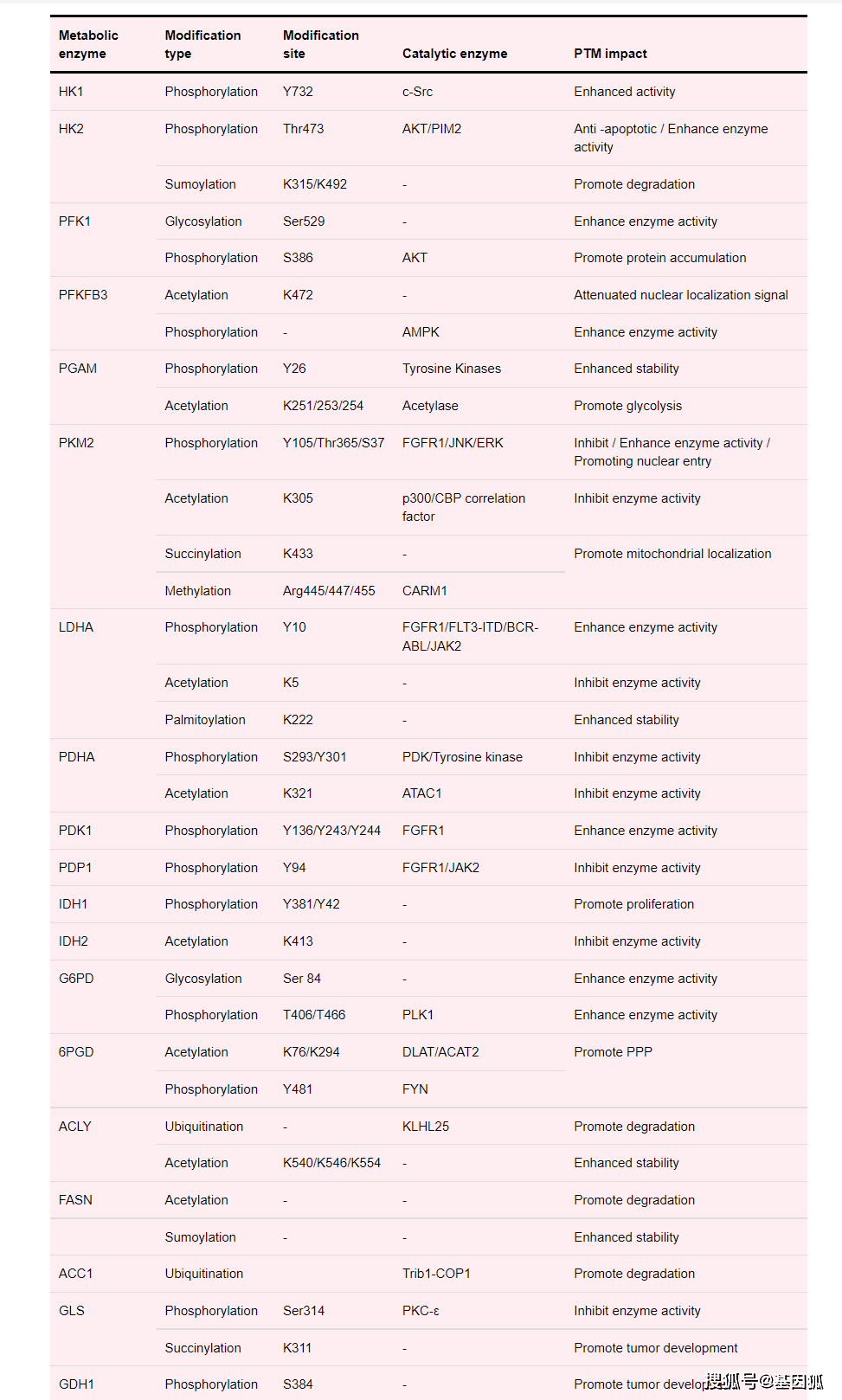
图1和表1分别归纳了肿瘤代谢中蛋白质翻译后修饰对代谢酶的调控和代谢酶翻译后修饰。
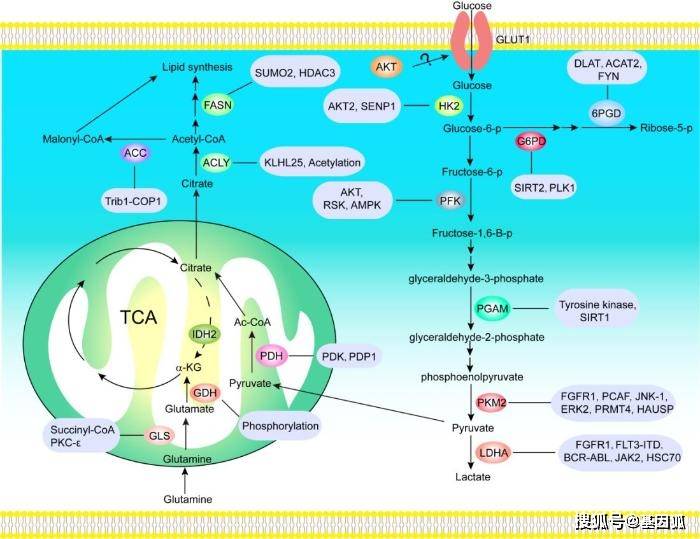 图1 肿瘤代谢中蛋白质翻译后修饰对代谢酶的调控
图1 肿瘤代谢中蛋白质翻译后修饰对代谢酶的调控
表1 代谢酶翻译后修饰
2 翻译后修饰与肿瘤细胞脂代谢和氨基酸代谢重编程
在肿瘤细胞中,脂肪酸代谢对其生长及转移具有重要意义。在合成方面,脂肪酸可参与癌细胞膜上磷脂的结构合成与重要信号的转导;在分解方面,肿瘤细胞利用脂肪酸β-氧化产生的ATP来维持所需的能量,以及利用烟酰胺腺嘌呤二核苷酸磷酸(NADPH)来维持体内的氧化还原平衡。因此,脂肪酸的合成和分解可能对肿瘤的发生和发展具有重要作用。柠檬酸裂解酶(adenosine triphosphate citrate lyase, ACLY)、乙酰CoA羧化酶(acetyl-CoA carboxylase, ACC)和脂肪酸合成酶(fatty acid synthetase, FASN) 串联活化的合成代谢过程。ACLY催化柠檬酸转化为草酰乙酸和乙酰CoA。
研究发现,CUL3通过其接合蛋白KLHL25与ACLY相互作用,使ACLY泛素化并降解,从而抑制脂肪酸从头合成。而ACLY在K540、K546、K554位点上发生乙酰化修饰后抑制ACLY泛素化修饰和降解,增强其稳定性,促进脂肪酸的从头合成,细胞增殖和肿瘤的生长。脂肪酸合成通路中的脂肪酸合成酶FASN (fatty acid synthase)也受乙酰化调控。FASN的乙酰化促进其通过泛素酶体系统降解,进一步减少脂质合成和肿瘤的生长。肝癌组织中由于FASN的表达增高,同时其去乙酰化酶HDAC3的表达增高,FASN乙酰化水平明显降低,与肝癌的发生发展密切相关,也提示靶向调控肿瘤细胞FASN乙酰化是潜在的治疗策略。也有研究显示,shRNA介导的ACAT1消融和GNPAT乙酰化缺陷,导致FASN降解,抑制异种移植和DEN/ccl4诱导的HCC的脂质代谢和肿瘤进展。在乳腺癌中,FASN被类泛素化(SUMO化),当SUMO2沉默引起SUMO化缺失会降低了FASN蛋白的稳定性。另外,乙酰辅酶A羧化酶1 (acetyl-CoA carboxylase 1, ACC1)是脂肪酸合成的限速酶。在急性髓性白血病,ACC1作为Trib1-COP1复合体(一种泛素连接酶)的靶标;ACC1突变体,由于缺少trib1结合位点而对降解具有抗性,从而减弱Trib1-COP1复合体驱动的白血病的发生。
氨基酸是生物合成过程的碳源和氮源,广泛参与蛋白质、核酸、脂类等大分子合成。为满足肿瘤细胞的生存和增殖,多种氨基酸的代谢处于失调状态,并在肿瘤发生发展中发挥重要作用。
谷氨酰胺是除葡萄糖外另一种被肿瘤细胞大量摄取的营养物质,主要通过以下三种途径促进肿瘤的发生发展:
① 通过分解产生α-酮戊二酸,进入三羧酸循环中代谢而提供能量;
② 提供生物大分子合成所必需的原料;
③ 产生还原型谷胱甘肽、NADPH,用以抵抗肿瘤细胞内高水平的活性氧。
作为谷氨酰胺分解代谢的第一个酶,谷氨酰胺酶(glutaminase, GLS)由琥珀酰辅酶A直接介导,在K311位点上发生琥珀酰化修饰,该修饰促进了GLS由单体向有活性的四聚体的转换,提高了其催化活性,增强了对谷氨酰胺的分解代谢。激活的GLS增加谷氨酰胺分解和还原性物质的产生,从而对抗氧化应激,促进肿瘤细胞存活和肿瘤生长。在临床样本中也发现,GLS K311琥珀酰化水平呈正相关,与胰腺导管腺癌患者的临床分期和不良预后呈正相关[47]。肾型谷氨酰胺酶(KGA)对肿瘤的代谢也很重要,其具有两种剪接变体谷氨酰胺酶C(glutaminase C, GAC)和谷氨酰胺酶M(GAM)。致癌蛋白Rho-C调节的PKC-ε激酶使GAC的Ser314处磷酸化而导致GAC活性升高;相反,KGA N末端区域的Ser95位点磷酸化便导致KGA活性降低,从而对肿瘤细胞的发生发展进行调控[48-49]。谷氨酰胺脱氢酶1 (GDH1)催化谷氨酸生成α-酮戊二酸,其S384位点的磷酸化与人胶质母细胞瘤的恶性程度及预后相关[50]。
3 小结与展望
越来越多的证据显示翻译后修饰对于肿瘤代谢重编程,发挥着重要的不可替代的作用。相对于包括Myc和HIF-1/2激活在内的作用比较缓慢的转录调控,翻译后修饰对代谢的调节迅捷而灵活。多种翻译后修饰对代谢酶的调节作用主要包括影响辅助因子的结合、底物结合、ATP的结合、复合物整体的形成、活性形式和非活性形式的转化、单体和多聚体的转化、代谢酶本身的降解,以及代谢酶在不同亚细胞器之间的转位等形式。另外,翻译后修饰的种类和形式也随着研究的深入逐步扩展,肿瘤细胞能够利用不同形式翻译后修饰之间存在的相互协同,相互竞争关系来为肿瘤的存活,增殖和转移提供生长优势。需要指出的是,由于篇幅的限制,本文只局限于肿瘤细胞本身翻译后修饰对代谢的重塑,对于肿瘤微环境中翻译后修饰如何通过代谢重编程,重新构建肿瘤细胞与免疫细胞,基质细胞等微环境中的通讯以及相互作用均没有涉及。今后的工作需要将蛋白组学、代谢组学、转录组学等多组学方法联合运用,全方位、多层次、多维度地揭示肿瘤发生发展的奥秘,为最终认识和治疗肿瘤奠定理论基础。
参考文献
范骏. 翻译后修饰与肿瘤代谢重编程. 中山大学学报(医学科学版)[J], 2022, 43(2): 161-172返回搜狐,查看更多
责任编辑: